Taaislijmziekte

Cystic fibrosis is een ziekte die ervoor zorgt dat dik, kleverig slijm zich ophoopt in de longen, het spijsverteringskanaal en andere delen van het lichaam. Het is een van de meest voorkomende chronische longziekten bij kinderen en jonge volwassenen. Het is een levensbedreigende aandoening.
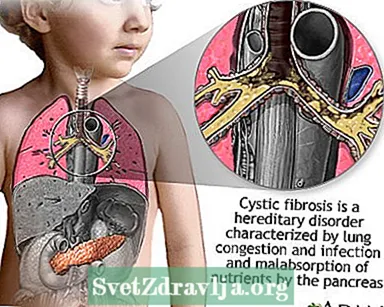
Cystic fibrosis (CF) is een ziekte die door families wordt doorgegeven. Het wordt veroorzaakt door een defect gen dat ervoor zorgt dat het lichaam abnormaal dikke en plakkerige vloeistof produceert, slijm genaamd. Dit slijm hoopt zich op in de luchtwegen van de longen en in de alvleesklier.
De ophoping van slijm resulteert in levensbedreigende longinfecties en ernstige spijsverteringsproblemen. De ziekte kan ook de zweetklieren en het voortplantingssysteem van een man aantasten.
Veel mensen dragen een CF-gen, maar hebben geen symptomen. Dit komt omdat een persoon met CF 2 defecte genen moet erven, 1 van elke ouder. Sommige Amerikanen hebben het CF-gen. Het komt vaker voor bij mensen van Noord- of Midden-Europese afkomst.
De meeste kinderen met CF worden gediagnosticeerd op de leeftijd van 2, vooral omdat screening van pasgeborenen in de Verenigde Staten wordt uitgevoerd. Bij een klein aantal wordt de ziekte pas ontdekt als ze 18 jaar of ouder zijn. Deze kinderen hebben vaak een mildere vorm van de ziekte.
Symptomen bij pasgeborenen kunnen zijn:
- Vertraagde groei
- Niet normaal aankomen tijdens de kindertijd
- Geen stoelgang in de eerste 24 tot 48 uur van het leven
- Zout smakende huid
Symptomen die verband houden met de darmfunctie kunnen zijn:
- Buikpijn door ernstige constipatie
- Verhoogd gas, een opgeblazen gevoel of een buik die gezwollen lijkt (opgezwollen)
- Misselijkheid en verlies van eetlust
- Krukken die bleek of kleikleurig zijn, stinken, slijm bevatten of drijven
- Gewichtsverlies
Symptomen die verband houden met de longen en sinussen kunnen zijn:
- Hoesten of toegenomen slijm in de sinussen of longen
- Vermoeidheid
- Neusverstopping veroorzaakt door neuspoliepen
- Herhaalde episodes van longontsteking (symptomen van longontsteking bij iemand met cystische fibrose zijn koorts, vaker hoesten en kortademigheid, meer slijm en verlies van eetlust)
- Sinuspijn of druk veroorzaakt door infectie of poliepen
Symptomen die later in het leven kunnen worden opgemerkt:
- Onvruchtbaarheid (bij mannen)
- Herhaalde ontsteking van de alvleesklier (pancreatitis)
- Ademhalingssymptomen
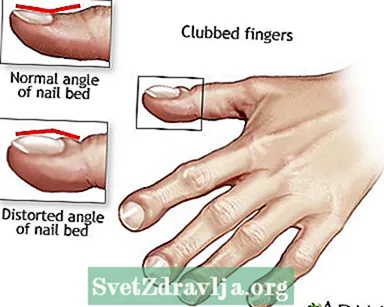
- Geknuppelde vingers
Er wordt een bloedonderzoek gedaan om CF te helpen opsporen. De test zoekt naar veranderingen in het CF-gen. Andere tests die worden gebruikt om CF te diagnosticeren, zijn onder meer:
- Immunoreactieve trypsinogeen (IRT)-test is een standaard screeningstest voor pasgeborenen voor CF. Een hoog niveau van IRT suggereert mogelijk CF en vereist verder testen.
- Zweetchloridetest is de standaard diagnostische test voor CF. Een hoog zoutgehalte in het zweet van de persoon is een teken van de ziekte.
Andere tests die problemen identificeren die verband kunnen houden met CF zijn onder meer:
- Röntgenfoto van de borst of CT-scan
- Fecale vettest
- Longfunctietesten
- Meting van de pancreasfunctie (ontlasting pancreas elastase)
- Secretine stimulatietest
- Trypsine en chymotrypsine in ontlasting
- Bovenste GI en dunne darm serie
- Longculturen (verkregen door sputum, bronchoscopie of keeluitstrijkje)
Een vroege diagnose van CF en een behandelplan kunnen zowel de overleving als de kwaliteit van leven verbeteren. Opvolging en monitoring zijn erg belangrijk. Indien mogelijk moet zorg worden verleend in een gespecialiseerde kliniek voor cystische fibrose. Wanneer kinderen de volwassen leeftijd bereiken, moeten ze overstappen naar een gespecialiseerd centrum voor cystische fibrose voor volwassenen.
Behandeling voor longproblemen omvat:
- Antibiotica om long- en sinusitis te voorkomen en te behandelen. Ze kunnen via de mond worden ingenomen, in de aderen of via ademhalingsbehandelingen worden toegediend. Mensen met CF kunnen antibiotica alleen gebruiken als dat nodig is, of altijd. Doses zijn vaak hoger dan normaal.
- Geneesmiddelen voor inhalatie om de luchtwegen te helpen openen.
- Andere geneesmiddelen die bij een ademhalingsbehandeling worden gegeven om het slijm dunner te maken en het ophoesten te vergemakkelijken, zijn DNAse-enzymtherapie en hooggeconcentreerde zoutoplossingen (hypertone zoutoplossing).
- Griepvaccin en pneumokokkenpolysaccharidevaccin (PPV) jaarlijks (vraag uw zorgverzekeraar).
- Longtransplantatie is in sommige gevallen een optie.
- Zuurstoftherapie kan nodig zijn als de longziekte erger wordt.
Longproblemen worden ook behandeld met therapieën om het slijm te verdunnen. Dit maakt het gemakkelijker om het slijm uit de longen te hoesten.
Deze methoden omvatten:
- Activiteit of oefening waardoor u diep ademhaalt breathe
- Apparaten die overdag worden gebruikt om te veel slijm uit de luchtwegen te verwijderen
- Manuele borstpercussie (of borstfysiotherapie), waarbij een familielid of een therapeut lichtjes in de borst, rug en het gebied onder de armen klapt
Behandeling voor darm- en voedingsproblemen kan zijn:
- Een speciaal dieet met veel eiwitten en calorieën voor oudere kinderen en volwassenen
- Pancreasenzymen om vetten en eiwitten te helpen absorberen, die bij elke maaltijd worden ingenomen
- Vitaminesupplementen, vooral vitamine A, D, E en K
- Uw leverancier kan andere behandelingen adviseren als u zeer harde ontlasting heeft
Ivacaftor, lumacaftor, tezacaftor en elexacaftor zijn geneesmiddelen die bepaalde soorten CF behandelen.
- Ze verbeteren de functie van een van de defecte genen die CF veroorzaakt.
- Tot 90% van de patiënten met CF die in aanmerking komen voor een of meer van deze geneesmiddelen, alleen of in combinatie.
- Als gevolg hiervan is er minder ophoping van dik slijm in de longen. Andere CF-symptomen zijn ook verbeterd.
Zorg en toezicht thuis moeten het volgende omvatten:
- Vermijd rook, stof, vuil, dampen, huishoudelijke chemicaliën, rook van open haarden en schimmel of meeldauw.
- Veel vocht geven, vooral aan zuigelingen en kinderen bij warm weer, bij diarree of dunne ontlasting, of tijdens extra lichamelijke activiteit.
- 2 of 3 keer per week trainen. Zwemmen, joggen en fietsen zijn goede opties.
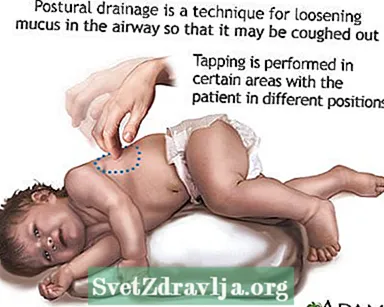
- Opruimen of naar boven halen van slijm of afscheidingen uit de luchtwegen. Dit moet 1 tot 4 keer per dag worden gedaan. Patiënten, families en zorgverleners moeten leren over borstpercussie en houdingsdrainage om de luchtwegen vrij te houden.
- Er wordt geen contact met andere mensen met CF aanbevolen omdat ze infecties kunnen uitwisselen (geldt niet voor familieleden).
U kunt de stress van ziekte verlichten door lid te worden van een ondersteuningsgroep voor cystische fibrose. Door te delen met anderen die gemeenschappelijke ervaringen en problemen hebben, kan uw gezin zich niet alleen voelen.
De meeste kinderen met CF blijven in goede gezondheid tot ze volwassen zijn. Ze kunnen aan de meeste activiteiten deelnemen en naar school gaan. Veel jonge volwassenen met CF maken hun studie af of vinden een baan.
Longziekte verergert uiteindelijk tot het punt waarop de persoon is uitgeschakeld. Tegenwoordig is de gemiddelde levensduur van mensen met CF die volwassen worden, ongeveer 44 jaar.
De dood wordt meestal veroorzaakt door longcomplicaties.
De meest voorkomende complicatie is een chronische luchtweginfectie.
Andere complicaties zijn onder meer:
- Darmproblemen, zoals galstenen, darmblokkade en rectale prolaps
- Bloed ophoesten
- Chronisch ademhalingsfalen
- suikerziekte
- Onvruchtbaarheid
- Leverziekte of leverfalen, pancreatitis, biliaire cirrose
- Ondervoeding
- Neuspoliepen en sinusitis
- Osteoporose en artritis
- Longontsteking die steeds terugkomt
- pneumothorax
- Rechtszijdig hartfalen (cor pulmonale)
- Colorectale kanker
Bel uw provider als een baby of kind symptomen van CF heeft en ervaringen heeft:
- Koorts, meer hoesten, veranderingen in sputum of bloed in sputum, verlies van eetlust of andere tekenen van longontsteking
- Verhoogd gewichtsverlies
- Frequentere stoelgang of ontlasting die stinkt of meer slijm heeft
- Gezwollen buik of toegenomen opgeblazen gevoel
Bel uw leverancier als een persoon met CF nieuwe symptomen ontwikkelt of als de symptomen verergeren, met name ernstige ademhalingsmoeilijkheden of bloed ophoesten.
CF is niet te voorkomen. Het screenen van mensen met een familiegeschiedenis van de ziekte kan het CF-gen in veel dragers detecteren.
CF
- Enterale voeding - kind - omgaan met problemen
- Gastrostomie voedingssonde - bolus
- Hoe ademen als je kortademig bent?
- Jejunostomie voedingssonde
- Houdingsdrainage
 Naar een discotheek gaan
Naar een discotheek gaan Houdingsdrainage
Houdingsdrainage Geknuppelde vingers
Geknuppelde vingers Taaislijmziekte
Taaislijmziekte
Donaldson SH, Pilewski JM, Griese M, et al. Tezacaftor/ivacaftor bij personen met cystische fibrose en F508del/F508del-CFTR of F508del/G551D-CFTR. Am J Respir Crit Care Med. 2018;197(2):214-224. PMID: 28930490 pubmed.ncbi.nlm.nih.gov/28930490/.
Eagan ME, Schechter MS, Voynow JA. Taaislijmziekte. In: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 21e ed. Philadelphia, PA: Elsevier; 2020: hoofdstuk 432.
Farrell PM, White TB, Ren CL, et al. Diagnose van cystische fibrose: consensusrichtlijnen van de Cystic Fibrosis Foundation. J Kinderarts. 2017;181S:S4-S15.e1. PMID: 28129811 pubmed.ncbi.nlm.nih.gov/28129811/.
Graeber SY, Dopfer C, Naehrlich L, et al. Effecten van lumacaftor/ivacaftor-therapie op de CFTR-functie bij homozygote Phe508del-patiënten met cystische fibrose. Am J Respir Crit Care Med. 2018;197(11)::1433-1442. PMID: 29327948 pubmed.ncbi.nlm.nih.gov/29327948/.
Grasemann H. Cystische fibrose. In: Goldman L, Schafer AI, eds. Goldman-Cecil Geneeskunde. 26e ed. Philadelphia, PA: Elsevier; 2020: hoofdstuk 83.
Rowe SM, Hoover W, Solomon GM, Sorscher EJ. Taaislijmziekte. In: Broaddus VC, Mason RJ, Ernst JD, et al, eds. Murray en Nadel's leerboek ademhalingsgeneeskunde. 6e druk. Philadelphia, PA: Elsevier Saunders; 2016: hoofdstuk 47.
Taylor-Cousar JL, Munck A, McKone EF, et al. Tezacaftor-ivacaftor bij patiënten met cystische fibrose die homozygoot is voor phe508del. N Engl J Med. 2017;377(21):2013-2023. PMID: 29099344 pubmed.ncbi.nlm.nih.gov/29099344/.
Zorg Ervoor Dat Je Eruit Ziet

Vervolg: Mijn angst voor vlees